




 Instagram
Instagram YouTube
YouTubeSupercomputers Help ID New Drug Leads to Fight Heart Disease
December 5, 2016
Contact: Kathy Kincade, kkincade@lbl.gov, +1 510 495 2124
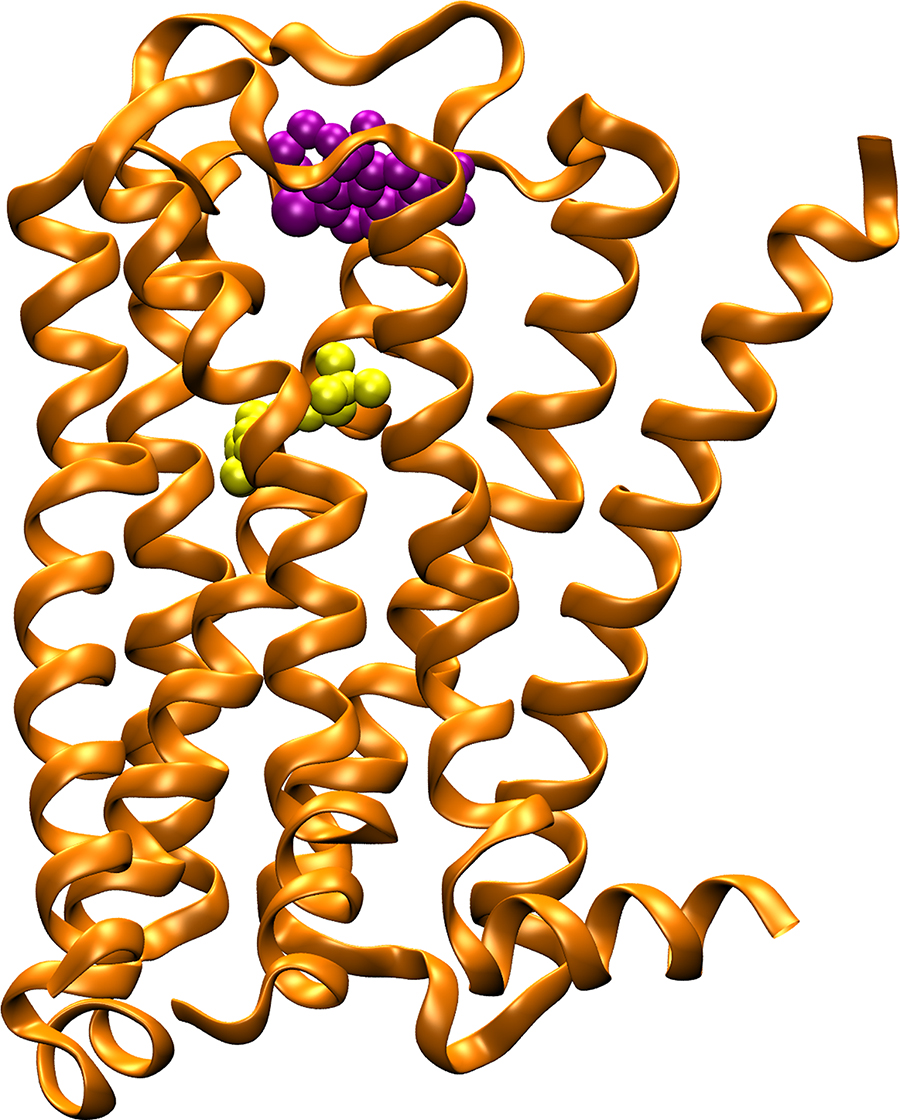
The M2 muscarinic acetylcholine receptor (orange ribbons) plays a key role in regulating the human heart rate and heart contraction forces. In contrast to compounds that bind to the highly conserved “orthosteric” binding site and cause serious side effects (yellow spheres), allosteric modulators (purple spheres) bind to target sites with great sequence diversity and provide potential selective treatment of heart disease. Image: Yinglong Miao, Howard Hughes Medical Institute
Using a unique computational approach to rapidly sample, in millisecond time intervals, proteins in their natural state of gyrating, bobbing and weaving, a research team from UC San Diego and Monash University in Australia has identified promising drug leads that may selectively combat heart disease, from arrhythmias to cardiac failure.
The researchers used supercomputing resources at the San Diego Supercomputer Center at UC San Diego, the Texas Advanced Computing Center at the University of Texas at Austin and the National Energy Research Scientific Computing Center (NERSC) at Berkeley Lab to perform an unprecedented survey of protein structures using accelerated molecular dynamics (aMD), a method that performs a more complete sampling of the myriad shapes and conformations that a target protein molecule may go through.
“The supercomputing power allows us to run hundreds of nanosecond aMD simulations, which are able to capture millisecond timescale events in complex biomolecules,” said the study’s first author, Yinglong Miao, a research specialist with the Howard Hughes Medical Institute at UC San Diego and research scientist with the UC San Diego Department of Pharmacology. “With our NERSC allocations, I was able to run a large portion of the aMD simulations to obtain enhanced sampling of our drug target, the M2 muscarinic G protein-coupled receptor. This helped us to capture the intrinsic flexibility of the receptor, which is key in molecular docking to identify the new drug lead compounds.”
Though effective in most cases, today’s heart medications—many of which act on M2 muscarinic acetylcholine receptors (M2 mAChRs) that decrease heart rate and reduce heart contractions—may carry side effects, sometimes serious. That’s because the genetic sequence of M2 mAChR’s primary orthosteric binding site is “highly conserved” and found in at least four other receptor types that are widely spread in the body, yielding unwanted results.
For this reason, drug designers are seeking a different approach, homing in on molecular targets or so-called “allosteric binding sites” that reside away from the receptor’s primary binding site and are built around a more diverse genetic sequence and structure. Essentially, allosteric modulators act as a kind of cellular dimmer-switch that, once turned on, ‘fine tunes’ the activation and pharmacological profile of the target receptor.
“Allosteric sites typically exhibit great sequence diversity and therefore present exciting new targets for designing selective therapeutics,” said the study’s co-investigator J. Andrew McCammon, the Joseph E. Mayer Chair of Theoretical Chemistry, a Howard Hughes Medical Institute investigator and Distinguished Professor of Pharmacology, all at UC San Diego.
In particular, drug designers have begun to aggressively search for allosteric modulators to fine-tune medications that bind to G protein-coupled receptors (GPCRs), the largest and most diverse group of membrane receptors in animals, plants, fungi and protozoa. These cell surface receptors act like an inbox for messages in the form of light energy, hormones and neurotransmitters, and perform an incredible array of functions in the human body.
In fact, between one-third to one-half of all marketed drugs act by binding to GPCRs, treating diseases including cancer, asthma, schizophrenia, Alzheimer’s and Parkinson’s disease and heart disease.
More Targeted Therapies
Though many of the GPCR drugs have made their way to the medicine cabinet, most—including M2 mAChR targeted drugs—exhibit side effects owing to their lack of specificity. All these drugs target the orthosteric binding sites of receptors, thus creating the push to find more targeted therapies based on allosteric sites.
“The problem here is that molecules that bind to these allosteric sites have proven extremely difficult to identify using conventional high-throughput screening techniques,” said McCammon.
Enter accelerated molecular dynamics and supercomputing. As described in the PNAS study, some 38 lead compounds were selected from a database of compounds from the National Cancer Institute, using computationally enhanced simulations to account for binding strength and receptor flexibility. About half of these compounds exhibited the hallmarks of an allosteric behavior in subsequent in vitro experiments, with about a dozen showing strong affinity to the M2 mAChR binding site. Of these, the researchers highlighted two showing both strong affinity and high selectivity in studies of cellular behavior. These cutting-edge experiments were performed by collaborators from the Monash Institute of Pharmaceutical Sciences.
The next steps will involve an investigation of the chemical properties of these novel molecules by the molecular chemists from Monash, led by Celine Valant and her colleague Arthur Christopoulos.
“This is just the beginning. We believe that it will be possible to apply our combined cutting-edge in silico and in vitro techniques to a wide array of receptor targets that are involved in some of the most devastating diseases,” said Valant.
This article is based on a news release provided by the University of California, San Diego.
About Computing Sciences at Berkeley Lab
High performance computing plays a critical role in scientific discovery. Researchers increasingly rely on advances in computer science, mathematics, computational science, data science, and large-scale computing and networking to increase our understanding of ourselves, our planet, and our universe. Berkeley Lab’s Computing Sciences Area researches, develops, and deploys new foundations, tools, and technologies to meet these needs and to advance research across a broad range of scientific disciplines.