




 Instagram
Instagram YouTube
YouTubeDOE CSGF Practicum Profile: Julianne Chung
Lawrence Livermore National Laboratory, 2007 "Practicum Proves Profitable for Image Reconstruction" An edited excerpt from DEIXIS (2008-2009), the DOE CSGF Annual
December 23, 2008
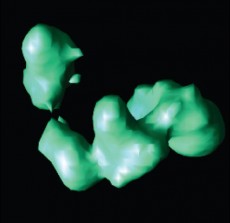
Isosurface rendering of a reconstructed TFIID protein using the new Lanczos-based iterative algorithm
Julianne Chung has an appreciation for versatility, starting with her academic interests. She showed an aptitude for mathematics at an all-girls high school in her hometown of Chattanooga, Tenn., but Chung has a love for dancing that dates back even further. She followed both interests and graduated in 2004 from Atlanta’s Emory University with a major in mathematics and a minor in dance and movement studies.
Versatility also attracted Chung to applied mathematics and computational science. Emory Mathematics and Computer Science Professor James Nagy suggested she do her honors thesis on image filtering algorithms. He showed her how they’re applicable to everything from astronomy to microscopy. Chung followed Nagy’s suggestion, then stayed on for graduate school at Emory so she could work with him.
Image reconstruction, especially from complex sources like medical PET and CAT scans, often requires the capability only high-performance computers can supply, Nagy says. Each image can contain huge amounts of data and sometimes several must be compared and averaged to create clearer, more detailed pictures out of motion blur and background noise.
Nagy recognized that Chung’s research had interesting parallels with a project involving Chao Yang, a staff scientist in Lawrence Berkeley National Laboratory’s computational research division.
Yang and his group were working with biochemists to develop algorithms that reconstruct images of complex particles like proteins and viruses.
The images come from a process called single-particle cryo-electron microscopy. In cryo-EM, as it’s called, a tiny purified sample of a biological substance is suspended in a water solution, then flash-frozen in liquid ethane. The sample is placed in the electron microscope, yielding thousands of two-dimensional images of single particles.
But scientists must set the microscope’s electron beam at a low level to avoid damaging the sample, so “What you get … is very fuzzy and low-contrast. The signal-to-noise ratio is very low,” Yang says. Proteins also can fold into many different conformations, making their structure look different from particle to particle, and the images show molecules oriented in random directions, like jacks scattered across the floor. “You have to determine the orientation (and) three-dimensional structure simultaneously,” Yang says.
Lastly, the images must be analyzed to find the particle’s “signature” and decipher its structure from the many varied orientations. It’s a computationally intensive job, Yang says, because up to 1 million images must be analyzed to achieve atomic-level resolution, and each image can have as many as 1 million pixels of data.
Yang and his group work on algorithms that accelerate image reconstruction. Their technique chooses a few good images, groups them according to their apparent orientations and averages them to mine the molecule’s shape from amid the noise. The information is used to make preliminary three-dimensional “seed” reconstructions.
Each two-dimensional image is compared with the seed reconstructions and grouped according to how much they differ from them. “Once you put each image into a different ‘bucket,’ each bucket is associated with a different orientation parameter,” Yang says. The algorithm then merges images in each group.
A multi-reference reconstruction algorithm continually updates each three-dimensional seed structure based on two-dimensional images that correspond to its orientation. After the structures are updated, the algorithm recalculates how much each two-dimensional image differs from them and regroups the images. The process is repeated until the two-dimensional images no longer change groups or a maximum number of iterations is reached.
Chung’s job, when she arrived at the Berkeley lab for her practicum in summer 2007, was to improve the algorithm’s speed and accuracy.
Building the seed structures for large proteins or viruses is the most computer-intensive step, Chung says. “This huge, 3-D volume has to reside on the memory of every single processor and the memory requirement is way too much.”
The group’s algorithm parceled out the two-dimensional images over many processors. Imagine the processors spread out in a single column and the images divided among the processors.
Chung added another level of parallelization by organizing the processors into a two-dimensional grid with multiple columns. That allowed her to partition the three-dimensional volume among multiple processors along with the two-dimensional images, cutting the memory demand. “The whole volume didn’t have to be on every processor,” Chung adds.
She also realized she could improve the cryo-EM implementation with research she did at Emory on regularization, which is designed to control errors in image reconstruction algorithms.
With inverse problems like image reconstruction, Yang says, “if you choose a very fast algorithm you may think it’s converging rapidly” on a solution. In fact, “As the algorithm moves along, the noise tends to be amplified.” To avoid that, the Berkeley group chose a slow algorithm and ran it for as many as 100 iterations.
Chung’s solution was to combine the Lanczos bidiagonalization algorithm – a standard iterative method – with a filtered singular value decomposition (SVD) approach. In essence, Yang says, the Lanczos method projects the big problem into a subspace to make the problem smaller. The projected problem then is solved with appropriate regularization by the SVD approach, a technique that is highly effective for small problems.
Chung tested her implementation on Jacquard, the 712-processor cluster at Berkeley Lab’s National Energy Research Scientific Computing Center (NERSC) and found that errors stabilize rather than “blowing up” into garbage. Yang says the number of iterations needed to reconstruct a three-dimensional protein image dropped to just 30. The improvements mean researchers can reconstruct high-resolution images of large structures like viruses.
“Julianne made a tremendous contribution to our project,” Yang says, not just improving algorithms but also implementing them. “That’s usually rare to do in three months.”
For her part, Chung loved her practicum: “To be able to … have a real-life application for something I was working on – that was a neat experience.”
About Computing Sciences at Berkeley Lab
High performance computing plays a critical role in scientific discovery. Researchers increasingly rely on advances in computer science, mathematics, computational science, data science, and large-scale computing and networking to increase our understanding of ourselves, our planet, and our universe. Berkeley Lab’s Computing Sciences Area researches, develops, and deploys new foundations, tools, and technologies to meet these needs and to advance research across a broad range of scientific disciplines.