




 Instagram
Instagram YouTube
YouTubeModeling Feat Sheds Light on Protein Channel's Function
November 1, 2012
NERSC Contact: Linda Vu, lvu@lbl.gov, +1 510 495 2402
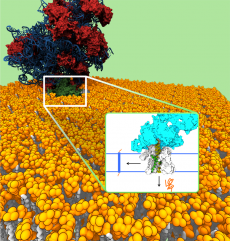
The ribosome (red-blue) in complex with the translocon channel (green), which is embedded in the cell membrane (yellow, white). Proteins that are inserted via the ribosome into the channel can either be laterally integrated into the cell membrane or secreted across the cell membrane (inset). (Image credit: Bin Zhang and Thomas Miller, Caltech 2012)
Using supercomputers at the Department of Energy’s National Energy Research Scientific Computing Center (NERSC), chemists have managed, for the first time, to simulate the biological function of a channel called the Sec translocon, which allows specific proteins to pass through membranes. The feat required bridging timescales from the realm of nanoseconds all the way up to full minutes, exceeding the scope of earlier simulation efforts by more than six orders of magnitude. The result is a detailed molecular understanding of how the translocon works.
Modeling behavior across very different timescales is a major challenge in modern simulation research. “We’ve created a tool that can actually be compared against experiments and even push experiments—to predict things that they haven’t been able to see,” says Thomas Miller, an assistant professor of chemistry at the California Institute of Technology (Caltech).
The new computational model and the findings based on its results are described by Miller and graduate student Bin Zhang in a recent issue of the journal Cell Reports.
Protein Gatekeepers in Action
The Sec translocon is a channel in cellular membranes involved in the targeting and delivery of newly made proteins. Such channels are needed because the proteins that are synthesized at ribosomes must travel to other regions of the cell or outside the cell in order to perform their functions; however, the cellular membranes prevent even the smallest of molecules, including water, from passing through them willy-nilly. In many ways, channels such as the Sec translocon serve as gatekeepers—once the Sec translocon determines that a given protein should be allowed to pass through, it opens up and allows the protein to do one of two things: to be integrated into the membrane, or to be secreted completely out of the cell.
Scientists have disagreed about how the fate of a given protein entering the translocon is determined. Based on experimental evidence, some have argued that a protein's amino-acid sequence is what matters—that is, how many of its amino acids interact favorably with water and how many clash. This argument treats the process as one in equilibrium, where the extremely slow rate at which a ribosome adds proteins to the channel can be considered infinitely slow. Other researchers have shown that slowing down the rate of protein insertion into the channel actually changes the outcome, suggesting that kinetic effects can also play a role.
"There was this equilibrium picture, suggesting that only the protein sequence is really important. And then there was an alternative picture, suggesting that kinetic effects are critical to understanding the translocon," Miller says. "So we wondered, could both pictures, in some sense, be right? And that turns out to be the case."
Computers Crack the Case
In 2010 and earlier this year, Miller and Caltech graduate student Bin Zhang published papers in the Proceedings of the National Academy of Sciences and the Journal of the American Chemical Society describing atomistic simulations of the Sec translocon. These computer simulations attempt to account for every motion of every single atom in a system—and typically require so much computing time that they can only model millionths of seconds of activity, at most. Meanwhile, actual biological processes involving proteins in the translocon last many seconds or minutes.
Miller and Zhang were able to use the atomistic simulations that they produced with NERSC resources to determine which parts of the translocon are most important and to calculate how much energy it costs those parts to move in ways that allow proteins to pass through. In this way, they were able to build a simpler version of the simulation that modeled important groupings of atoms, rather than each individual atom. Using the simplified simulation, they could simulate the translocon's activity over the course of more than a minute.
Using the simplified model, Miller and his team observed the different ways in which proteins move through the channel. In the simulation, any number of variables could be changed—including the protein's amino-acid sequence, its electronic charge, the rate at which it is inserted into the translocon, the length of its tail, and more. The effect of these alterations on the protein's fate was then studied, revealing that proteins move so slowly within the tightly confined environment of the translocon that the pace at which they are added to the channel during translation—a process that might seem infinitely slow—can become important. At the same time, Miller and Zhang saw that other relatively fast processes give rise to the results associated with the equilibrium behavior.
"In fact, both equilibrium and kinetically controlled processes are happening—but in a way that was not obvious until we could actually see everything working together," Miller says. “NERSC resources were the primary driving force in making this research possible.”
He notes that even the simplified model was really computationally expensive. “It took between four and forty hours on a single processor to compute each trajectory for the coarse-grained system,” says Miller. “In total, we computed over 200,000 trajectories.”
Beyond elucidating how the translocon works and reconciling seemingly disparate experimental results, the new simulation also lets the researchers perform experiments computationally that have yet to be tried in the lab. For example, they have run simulations with longer proteins and observed that at such lengths—unlike what has been seen with shorter proteins—the equilibrium picture begins to be affected by kinetic effects.
"This could bring the two experimental camps together, and to have led that would be kind of exciting," Miller says.
The new Cell Reports paper is titled "Long-timescale dynamics and regulation of Sec-facilitated protein translocation." The work was supported by the U.S. Office of Naval Research and the Alfred P. Sloan Foundation, with computational resources provided by the U.S. Department of Energy, the National Science Foundation, and the National Institute of General Medical Sciences.
This story was adapted from a press release issued by Caltech.
About Computing Sciences at Berkeley Lab
High performance computing plays a critical role in scientific discovery. Researchers increasingly rely on advances in computer science, mathematics, computational science, data science, and large-scale computing and networking to increase our understanding of ourselves, our planet, and our universe. Berkeley Lab’s Computing Sciences Area researches, develops, and deploys new foundations, tools, and technologies to meet these needs and to advance research across a broad range of scientific disciplines.