




 Instagram
Instagram YouTube
YouTubeNew Mathematical Method Reveals Where Genes Switch On or Off
“Compressed sensing” determines atomic-level energy potentials with accuracy approaching experimental measurement
February 22, 2012
John Hules, JAHules@lbl.gov, +1 510 486 6008
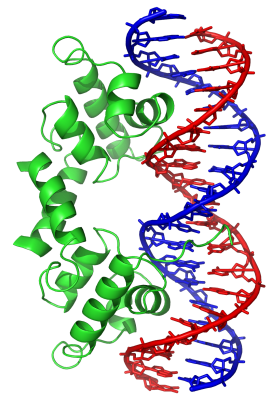
Figure 1. Helix-turn-helix (HTH) proteins are the most widely distributed family of DNA-binding proteins, occurring in all biological kingdoms. This image shows a lambda repressor HTH transcription factor (green) binding to a lambda operator DNA sequence (blue and red) of the virus bacteriophage lambda. Image: Richard Wheeler, Wikipedia
Developmental biologists at Stanford University, using computing resources at the U.S. Department of Energy’s National Energy Research Scientific Computing Center (NERSC), have taken a new mathematical method used in signal processing and applied it to biochemistry, using it to reveal the atomic-level details of protein–DNA interactions with unprecedented accuracy. They hope this method, called “compressed sensing,” will speed up research into where genes are turned on and off, and they expect it to have applications in many other scientific domains as well.
“Proteins are what my dreams are made of—this is true of everyone,” says Mohammed AlQuraishi, a postdoctoral researcher in the Developmental Biology Department at Stanford. “Proteins are also what my dreams are about—in this I am a little more peculiar. I dream about proteins because I’m often thinking about them, and I’m often thinking about them because I find them interesting. And part of what interests me about proteins is their capacity for gene regulation.”
All of the cells in an individual human body contain exactly the same DNA, which provides the blueprint for building and operating a complete organism. Yet cells in different organs have different structures and perform different functions. “What distinguishes between a cell in the eye as opposed to a cell in the elbow are the subset of genes that are expressed in those cells, and what determines that are the transcription factors—they’re the programming of the cell,” AlQuraishi explains.
Transcription factors are proteins that bind to specific DNA sequences to either activate or block the expression of a gene (Figure 1). “The transcription factors determine what’s on and what’s off at any given point in time and space,” AlQuraishi says. There are approximately 2000 human transcription factors, making them the single largest family of human proteins. They are involved in many important processes, including cellular differentiation, responses to intercellular signals, responses to environmental stimuli, cell growth and death, the onset of certain diseases, especially cancers, and responses to certain drugs.
Understanding exactly where transcription factors bind with DNA could help answer many crucial questions in biology. But determining this experimentally requires significant labor and financial resources, so a computational solution is highly desirable.
Analyzing biochemical phenomena at the molecular level involves determining the energy potential, a mathematical description of the energy of every possible interaction in a molecular system. Currently it’s not feasible to calculate energy potentials for large molecular systems from first principles, even on supercomputers—the calculations are simply too large. Instead, potentials are inferred from a mixture of theoretical modeling and experimental data, using either physical or statistical models. But the models are imprecise, and the theoretical development of some potentials has taken decades. The accuracy of both laboratory and computational studies of phenomena ranging from pharmaceutical drug interactions to protein folding to gene expression is often limited by the accuracy of the energy potentials.
Compressed sensing: from information theory to biology
 and compressed sensing images (right) from Rice University’s one-pixel camera using 1600 measurements. Images: Digital Signal Processing Group, Rice University")
Figure 2. Original images (left) and compressed sensing images (right) from Rice University’s one-pixel camera using 1600 measurements. Images: Digital Signal Processing Group, Rice University
Fortunately, AlQuraishi has other interests besides proteins, including machine learning, mathematics, and computer languages, and he’s not afraid to mix these fields together. It was in a statistics class at Stanford—“Compressed Sensing,” taught by a co-founder of this new field, Emmanuel Candès—that AlQuraishi found a new and unexpected way to determine energy potentials.
Compressed sensing is the recovery of sparse signals from a few carefully constructed but seemingly random measurements. (In mathematical terms, it’s a technique for finding sparse solutions to underdetermined linear systems.) When the first papers on compressed sensing were published less than a decade ago, it was quickly recognized as a breakthrough in information theory, because it allowed a complete signal (such as a visual image) to be communicated and reconstructed with much less data than previously thought possible.
The first uses of compressed sensing were in electrical engineering and signal processing, such as MRI signal processing. With compressed sensing, an accurate medical MRI image can be constructed from a smaller number of data points, allowing the patient to spend less time in the MRI chamber.
In another application, researchers at Rice University used compressed sensing to create a one-pixel camera that produces slightly blurry but easily recognizable photos (Figure 2). Compressed sensing algorithms allow the image to be reconstructed from the sum of the values of a small number of pseudorandom samples. Since the camera relies on a single photon detector, it can be adapted for use at wavelengths where conventional scientific optical detectors are blind.
AlQuraishi’s “aha” moment came when he realized that determining atomic-level energy potentials in protein–DNA complexes could be treated mathematically as a signal acquisition problem. By using the crystal structures of the complexes as the “camera” or sensors, and using the experimentally determined binding affinities of the complexes as the compressive measurements, he could determine the “signal”—the attraction between specific pairs of atoms in the protein and DNA. The crystal structures are typically obtained by X-ray crystallography or nuclear magnetic resonance (NMR) spectroscopy and are archived in the Protein Data Bank, a community resource for biological research.
“Measurements that we take for granted as being one kind of measurement, a structural measurement, can actually be seen as a different kind of measurement,” AlQuraishi explains. “In a sense it’s two different mathematical formulations, one that’s of use for the structural stuff, and one that’s used for the statistical compressed sensing stuff. On face value they look different. But I had been staring at each individually for a long time, and it occurred to me that with a simple transformation, you could get one to look like the other.”
AlQuraishi’s advisor and collaborator at Stanford, Harley McAdams, a research professor of developmental biology, explains why this transformation works: “You have to ask the question, Why does a protein bind to DNA? It’s because there’s a set of atoms in the protein and a set of atoms in the DNA that have an attraction to each other—that’s what we call a potential. The sum of these individual atomic attractions gives you the net binding energy of the protein to the DNA. If you were able to figure out the set of all atomic relationships or proximities within this structure—which you can from the crystal structures, and that’s where a lot of computation comes in—and you did that for a lot of different cases, then you could statistically determine which of these interactions are important.”
That’s exactly what the compressed sensing computation does, says McAdams: “Given what we know about the set of atomic proximities between the protein and the DNA from these different cases, and what we know about their binding energies, we can take that information and infer what are the atom-to-atom potentials.”
AlQuraishi sums it up by saying, “The idea is that these protein–DNA complexes serve as natural experimental apparatus, because we know where the two pieces, the protein and DNA, are; and we also know the energy of that interaction. So each complex is effectively a probe into the underlying biophysics of protein–DNA interactions at the atomic level.”
“And it’s a direct measurement of the atomic-level interactions,” McAdams adds. “All other previous methods have not been that direct—they’ve been just purely statistical correlations, or based upon some hypothesized physical theory that would be applicable. But this method doesn’t use any hypothetical theory, it says OK, let’s just go in and measure it. That’s its power.”
AlQuraishi and McAdams call this method de novo potential determination, meaning it’s done from scratch, with no assumptions. Figure 3 is a schematic representation of how the method works.
 to attractive (dark blue). The interatomic protein–DNA potential can be inferred by ℓ1 minimization from measurements provided by a small number of sensors (protein–DNA structures + binding energies). Image: Mohammed AlQuraishi and Harley McAdamsFigure 3. De novo potential determination as an application of compressed sensing. Select image to enlarge. The potential is represented by a heat map of microscopic interaction energies ranging from repulsive (dark pink) to attractive (dark blue). The interatomic protein–DNA potential can be inferred by ℓ1 minimization from measurements provided by a small number of sensors (protein–DNA structures + binding energies). Image: Mohammed AlQuraishi and Harley McAdams")
Figure 3. De novo potential determination as an application of compressed sensing. Select image to enlarge. The potential is represented by a heat map of microscopic interaction energies ranging from repulsive (dark pink) to attractive (dark blue). The interatomic protein–DNA potential can be inferred by ℓ1 minimization from measurements provided by a small number of sensors (protein–DNA structures + binding energies). Image: Mohammed AlQuraishi and Harley McAdams
To test this method, the two researchers used it to determine the DNA-binding affinity of 63 helix-turn-helix (HTH) proteins, the most widely distributed family of DNA-binding proteins, with a large number of structures in the Protein Data Bank. HTH proteins occur in all biological kingdoms; they include virtually all bacterial transcription factors and about 25 percent of human transcription factors. The results, published in the Proceedings of the National Academy of Sciences (PNAS), were approximately 90 percent accurate, compared to approximately 60 percent for the best performing alternative computational methods.[1]
Applications: from gene therapy to synthetic biology
AlQuraishi and McAdams hope the de novo potential method will accelerate research in transcription factors and gene expression. “To figure out where a transcription factor binds is a rather tedious operation in the lab. It takes a lot of time and money,” McAdams explains. “If we could figure that out computationally, it would enable us to cut to the chase and figure out the cell’s regulatory logic without doing all that experimental work, except for verification.”
They also hope that this method will be adopted by researchers in the Protein Structure Initiative (PSI), a federal, university, and industry effort to make the three-dimensional, atomic-level structures of most proteins easily obtainable. To date, there has been no systematic and widely agreed on method to select proteins for this kind of research. The de novo potential method “provides a principled means for deciding what additional structures would give you the most information for inferring atomic-level potentials,” McAdams points out. For example, if crystal structures had been available for 90 HTH proteins instead of only 63, the accuracy of the de novo potentials would have been even better.
“And why do you want the energy potentials to be accurate?” McAdams asks. “Well, these energy potentials are a starting point for a huge number of different types of biophysical calculations, including medically important enzymatic reactions, and metabolic pathways important in energy production and synthetic biology.”
“Energy potentials are very fundamental,” AlQuraishi adds. “Any kind of simulation—physics, chemistry, biology—has a potential underneath it that’s driving the whole thing.”
“The project also has practical applications, including gene therapy,” McAdams continues. “Gene therapy relies on using DNA-binding proteins to bind DNA and integrate new genetic material into the patient. The ability to predict protein-DNA interactions will enable the design of better gene therapy-based therapeutics.”
The researchers’ association with NERSC began with a 2010 NERSC Initiative for Scientific Exploration (NISE) award of 1,880,000 processor hours, which were run primarily on NERSC’s Carver system; 1,720,000 more hours were added during that year. Together, these computer runs—hundreds of independent simulations, each running on a separate processor—produced the findings reported in the PNAS article.
“To develop this model, we needed to explore a very large space of possible models, which was only possible with the computing resources at NERSC,” McAdams says.
[1] Mohammed AlQuraishi and Harley McAdams, “Direct inference of protein–DNA interactions using compressed sensing methods,” PNAS 108, 14819 (2011), doi: 10.1073/pnas.1106460108.
About Computing Sciences at Berkeley Lab
High performance computing plays a critical role in scientific discovery. Researchers increasingly rely on advances in computer science, mathematics, computational science, data science, and large-scale computing and networking to increase our understanding of ourselves, our planet, and our universe. Berkeley Lab’s Computing Sciences Area researches, develops, and deploys new foundations, tools, and technologies to meet these needs and to advance research across a broad range of scientific disciplines.