




 Instagram
Instagram YouTube
YouTubeMachine Learning Method Accurately Predicts Metallic Defects
Berkeley Lab Research Could Accelerate Development of New Metallic Materials for the Aerospace Industry
February 3, 2017
Linda Vu, lvu@lbl.gov, +1 510.495.2402
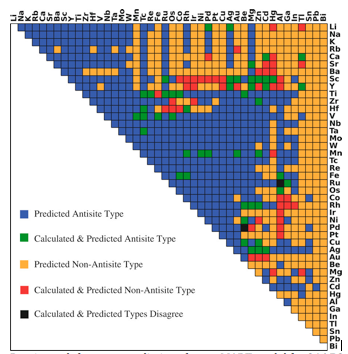
Dominant defect type predictions from r-MART model for 946 B2-type intermetallics. Colors indicates the relationship between prediction and calculations as shown in the legend.
For the first time, researchers at the Lawrence Berkeley National Laboratory (Berkeley Lab) have built and trained machine learning algorithms to predict defect behavior in certain intermetallic compounds with high accuracy. This method will accelerate research of new advanced alloys and lightweight new materials for applications spanning automotive to aerospace and much more.
Their results were published in the December 2016 issue of Nature Computational Materials.
Materials are never chemically pure and structurally flawless. They almost always contain defects, which play an important role in dictating properties. These defects may appear as vacancies, which are essentially ‘holes’ in the substance’s crystal structure, or antisite defects, which are essentially atoms placed on the wrong crystal site. Understanding of such point defects is crucial for scientists designing materials because they can have a dramatic effect on long-time structural stability and strength.
Traditionally, researchers have used a computational quantum mechanical method known as density functional calculations to predict what kinds of defects can be formed in a given structure and how they affect the material’s properties. Although effective, this approach is very computationally expensive to execute for point defects limiting the scope of such investigations.
“Density functional calculations work well if you are modeling one small unit, but if you want to make your modeling cell bigger the computational power required to do this increases substantially,” says Bharat Medasani, a former Berkeley Lab postdoc and lead author of the paper. “And because it is computationally expensive to model defects in a single material, doing this kind of brute force modeling for tens of thousands of materials is not feasible.”
To overcome these computing challenges, Medasani and his colleagues developed and trained machine learning algorithms to predict point defects in intermetallic compounds, focusing on the widely observed B2 crystal structure. Initially, they selected a sample of 100 of these compounds from the Materials Project Database and ran density functional calculations on supercomputers at the National Energy Research Scientific Computing Center (NERSC), a DOE Office of Science User Facility at Berkeley Lab, to identify their defects.
Because they had a small data sample to work from, Medasani and his team used a forest approach called gradient boosting to develop their machine learning method to a high accuracy. In this approach additional machine learning models were built successively and combined with prior models to minimize the difference between the models predictions and the results from density functional calculations. The researchers repeated the process until they achieved a high level of accuracy in their predictions.
“This work is essentially a proof of concept. It shows that we can run density functional calculations for a few hundred materials, then train machine learning algorithms to accurately predict point defects for a much larger group of materials,” says Medasani, who is now a postdoctoral researcher at the Pacific Northwest National Laboratory.
“The benefit of this work is now we have a computationally inexpensive machine learning approach that can quickly and accurately predict point defects in new intermetallic materials ” says Andrew Canning, a Berkeley Lab Computational Scientist and co-author on the paper. “We no longer have to run very costly first principle calculations to identify defect properties for every new metallic compound.”
“This tool enables us to predict metallic defects faster and robustly, which will in turn accelerate materials design,” says Kristin Persson, a Berkeley Lab Scientist and Director of the Materials Project, an initiative aimed at drastically reducing the time needed to invent new materials by providing open web-based access to computed information on known and predicted materials. As an extension of this work an open source Python toolkit for modeling point defects in semiconductors and insulators (PyCDT) has been developed.
In addition to Medasani, Canning and Persson, other authors on the NatureComputational Materials paper include: Hong Ding, Wei Chen, Mark Asta and Maciej Haranczyk (Berkeley Lab); and Anthony Gamst (University of California, San Diego). Additionally, Danny Broberg (University of California, Berkeley), Geoffroy Hautier (University Catholique de Louvain, Belgium) and Nils Zimmermann (Berkeley Lab) were involved in the development of the PyCDT software.
The research was supported by the Department of Energy’s Office of Science.
About Computing Sciences at Berkeley Lab
High performance computing plays a critical role in scientific discovery. Researchers increasingly rely on advances in computer science, mathematics, computational science, data science, and large-scale computing and networking to increase our understanding of ourselves, our planet, and our universe. Berkeley Lab’s Computing Sciences Area researches, develops, and deploys new foundations, tools, and technologies to meet these needs and to advance research across a broad range of scientific disciplines.