




 Instagram
Instagram YouTube
YouTubeMachine Learning Software that Enhances Molecular Dynamics Modeling Wins Gordon Bell Prize
CRD researchers are co-authors on a paper that won this prestigious award at SC20
November 19, 2020
Contact: cscomms@lbl.gov
Editor's Note: This story has been updated to reflect that the DeePMD-kit paper won the 2020 ACM Gordon Bell Prize. The award was announced at SC20 on Thursday, November 19.
Researchers from Lawrence Berkeley National Laboratory’s Computational Research Division (CRD) are co-authors on a research paper that has been awarded the 2020 ACM Gordon Bell Prize at SC20 for a new machine-learning-based software package that enhances molecular dynamics modeling.
The SC20 paper, “Pushing the limit of molecular dynamics with ab initio accuracy to 100 million atoms with machine learning,” describes DeePMD-kit (DeePMD stands for deep potential molecular dynamics), an optimized ab initio molecular dynamics (AIMD) code developed by an international team that includes Lin Lin, a CRD faculty scientist and UC Berkeley mathematics professor, and Weile Jia, a UC Berkeley postdoc who is a member of Lin’s research group. The project was partially funded by a Department of Energy’s Early Career Research Program award that Lin received in 2017. Jia and Linfeng Zhang – a recent postdoc who graduated from Princeton University – together led this project.
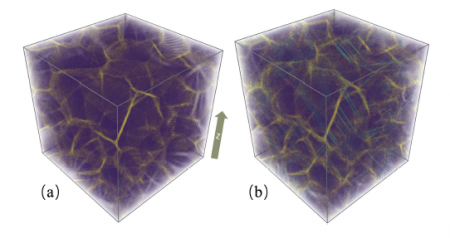
(a) A 10,401,218-atom nanocrystalline copper consisting of 64 randomly oriented crystals with 15-nm averaged grain diameter. (b) The nanocrystalline copper after 10 percent tensile deformation along the z-axis. Purple, yellow, and cyan denote the atoms in the grains, atoms in the grain boundaries, and atoms in the stacking faults. Credit: Weile Jia, et al
AIMD is a popular simulation tool for studying and describing atomic processes that occur in materials and molecules. While AIMD has long been the method of choice for modeling complex atomistic phenomena from first principles, it relies on computationally expensive electronic structure models such as density functional theory (DFT) and post-DFT theories. Latin for “from the start,” ab initio models calculate interactions between each atom in a model using the most basic laws of physics, sometimes referred to as “first principles calculations.” These models, however, consume an enormous amount of compute power.
“Most DFT-based AIMD calculations are performed with hundreds of atoms for up to a few picoseconds per day,” Lin said. In the past two decades, he added, significant progress has been made in pushing forward the spatial scale of electronic structure methods to study systems comprising up to millions of atoms at least for a certain class of materials. However, relatively little progress has been made in increasing the temporal scale (time scale) accessible to AIMD.
“In application areas such as materials science, chemistry, combustion, and biology-related systems like protein folding, AIMD is the basic tool for understanding atomic structures and atomic behaviors of these systems,” Zhang said. “But all these things are limited by their accuracy when we look at large systems, which current AIMD models cannot handle, even with the help of powerful supercomputers.”
Applying Neural Networks to ab initio Data
To address the grand challenge of modeling larger systems, DeePMD-kit uses a neural network to guide molecular dynamic calculations, Jia explained. By approximating the ab initio data with deep neural networks, DeePMD reduces the computational complexity from cubic to linear scaling, significantly boosting the efficiency. It also demonstrates what can be achieved by integrating physics-based modeling and simulation, machine learning, and efficient implementation on a next-generation computational platform.
Using this approach, DeePMD-kit has the potential to improve the productivity and accuracy of AIMD modeling across chemistry, biology, materials science, and other scientific disciplines, according to the researchers. In early testing, it has been used to model various phenomena in physical chemistry and materials science and demonstrated its ability to boost the spatial and temporal scale accessible by AIMD without losing ab initio accuracy.
For the SC20 paper, the research team ran DeePMD on the Summit supercomputer at the Oak Ridge Leadership Computing Facility (OLCF) on a test case of a block of copper atoms and was able to simulate systems with more than 100 million atoms and efficiently perform AIMD simulations for up to a few nanoseconds per day of supercomputer time. This increases the spatial scale by at least 100x compared to the largest system simulated by all previous Gordon Bell award-winning works, and reduces the time-to-solution by at least 1000x, Lin noted. By efficiently scaling the code up to the entire Summit supercomputer, they attained 91 PFLOPS in double precision (45.5% of the peak) and 162/275 PFLOPS in mixed-single/half precision. “This opens the door to simulating unprecedented size and time scales with ab initio accuracy,” the researchers write.
DeePMD-kit is open source and can be adapted to current and future computing platforms beyond CPUs and GPUs; several research groups are already working on projects related to this, according to Jia. The group is now restructuring DeePMD and merging aspects they modified for the current HPC project so that DeePMD-kit can be useful both for large-scale applications involving systems with 100 million atoms, as well as for applications at smaller scales when only a few GPUs are available. “The latter paradigm may be even more useful for many users,” Jia said.
“The good thing is that most of the techniques we used for thousands of GPUs (on Summit) also work for one or two GPUs, which means the GPU-enabled DeePMD-kit can be applied to a broad range of applications,” Zhang added.
In addition to Jia, Lin, and Zhang, co-authors on the SC20 paper include Han Wang (Institute of Applied Physics and Computational Mathematics, Beijing); Mohan Chen and Denghui Lu (Peking University, Beijing); and Roberto Car and Weinan E (Princeton University)
OLCF is DOE Office of Science user facility.
About Computing Sciences at Berkeley Lab
High performance computing plays a critical role in scientific discovery. Researchers increasingly rely on advances in computer science, mathematics, computational science, data science, and large-scale computing and networking to increase our understanding of ourselves, our planet, and our universe. Berkeley Lab’s Computing Sciences Area researches, develops, and deploys new foundations, tools, and technologies to meet these needs and to advance research across a broad range of scientific disciplines.